DFT Analysis of Hypercoordinate Phosphine Oxide-Containing Organotin (IV) Compounds



Siobhan Renee Liu, Dr. R. Stephen Wylie*
Foucher Group, Ryerson University
B.Sc. (Hons.) Thesis, 2019-2020My presentation for the 48th Southern Ontario Undergraduate Student Chemistry Conference (SOUSCC48) that I was never able to present because COVID-19 forced the conference to be cancelled. Presented with more text than usual since there's no audio to go with it.
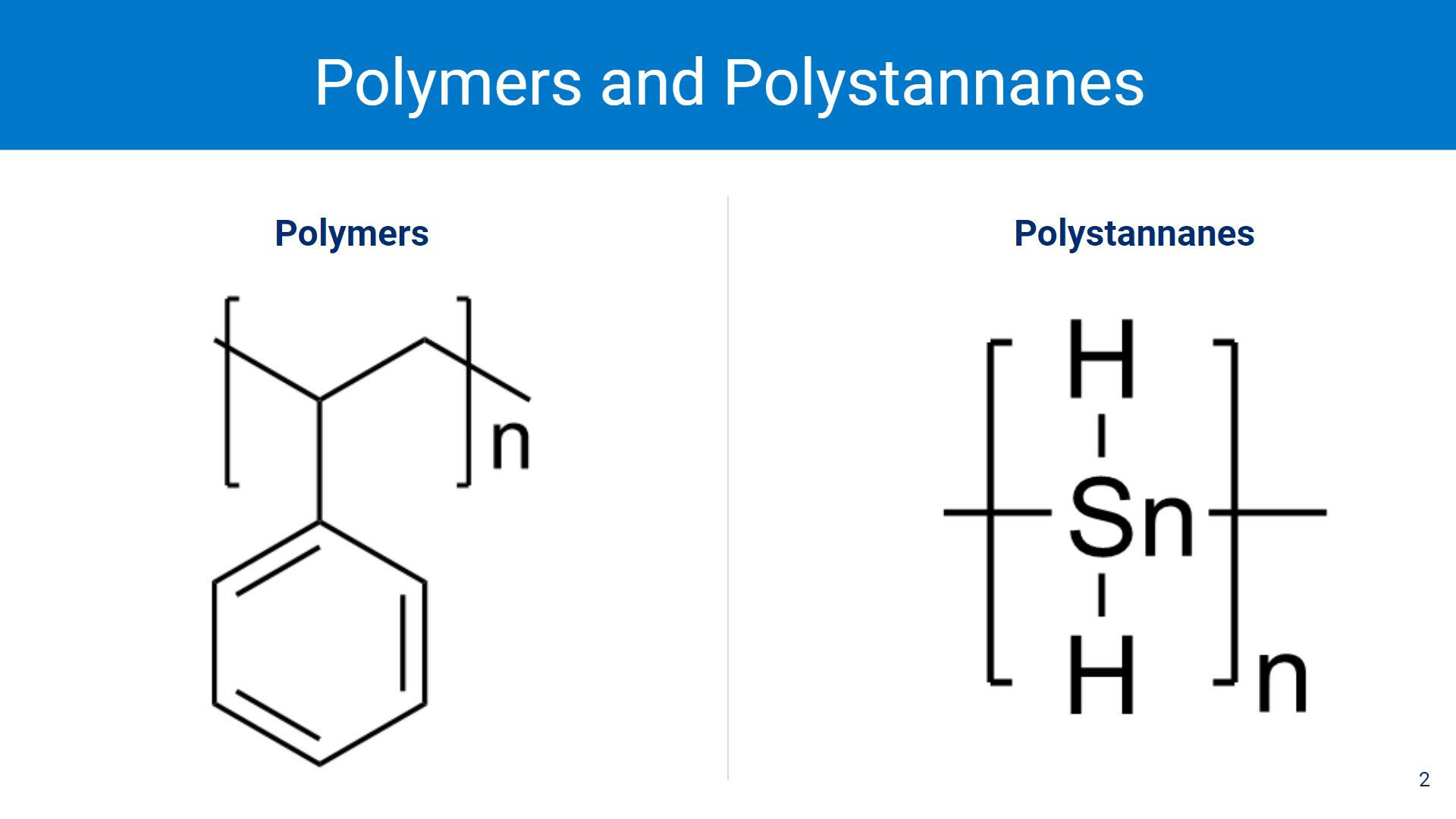
Polymers are a large macromolecule with repeating subunits called monomers. The most common use for polymers is in the development and manufacturing of plastics (polystyrene, pictured on the left, is used to make Styrofoam).Polystannanes (pictured on the right) are essentially polymers but with a repeating tin (Sn) backbone. Polystannanes have the potential to be semiconductors for electronic applications.
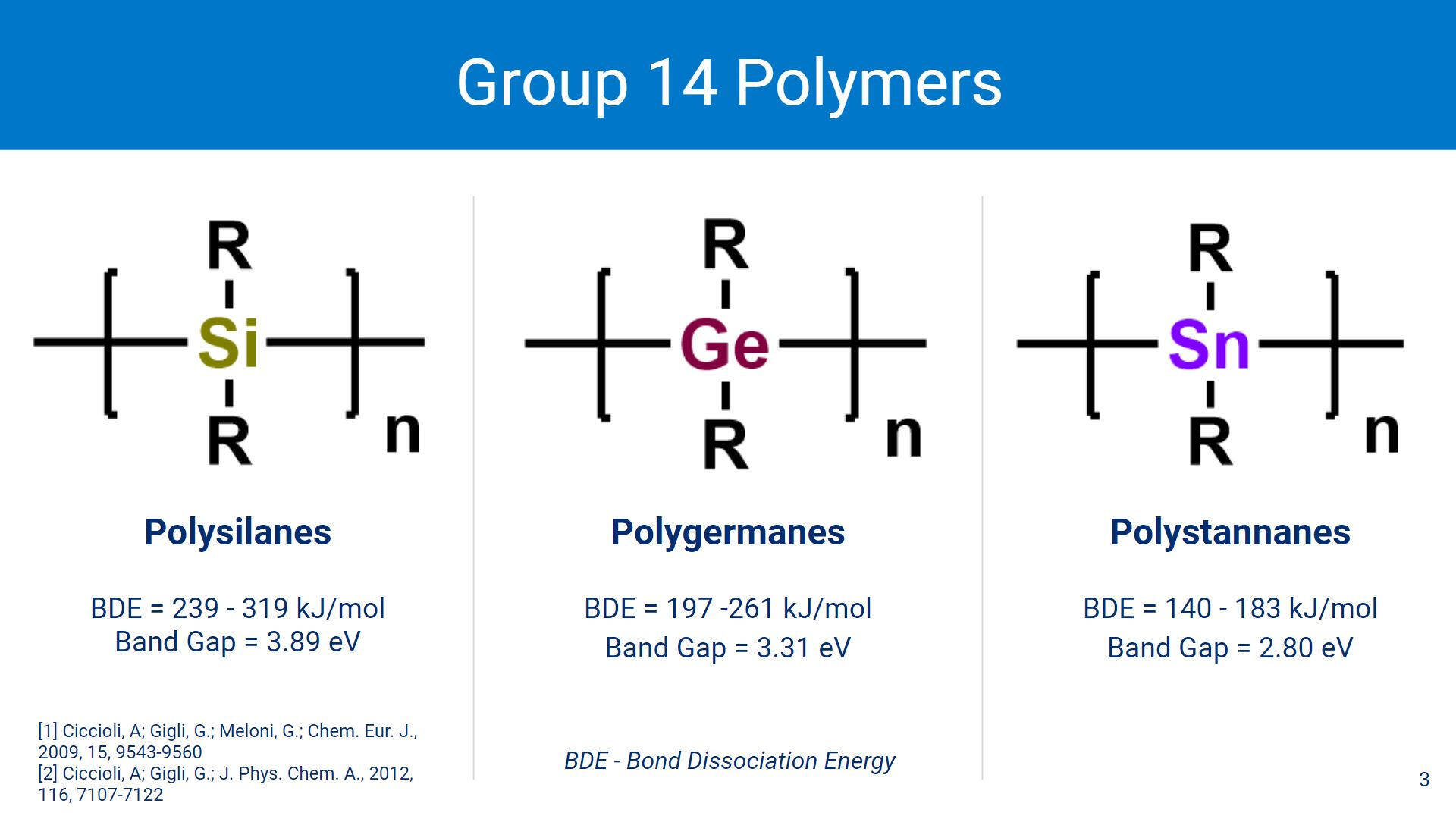
When compared to other Group 14 polymers, polystannanes are shown to have a smaller band gap (2.80 eV) compared to polysilanes (3.89 eV) and polygermanes (3.31 eV). A smaller band gap allows for better conductivity, but polystannanes also have the largest bond dissociation energy out of the 3 classes of polymers. A larger bond dissociation energy leads to weaker bonds between the Sn-Sn centers.
References:
[1] https://doi.org/10.1002/chem.200900804
[2] https://pubs.acs.org/doi/10.1021/jp300624z
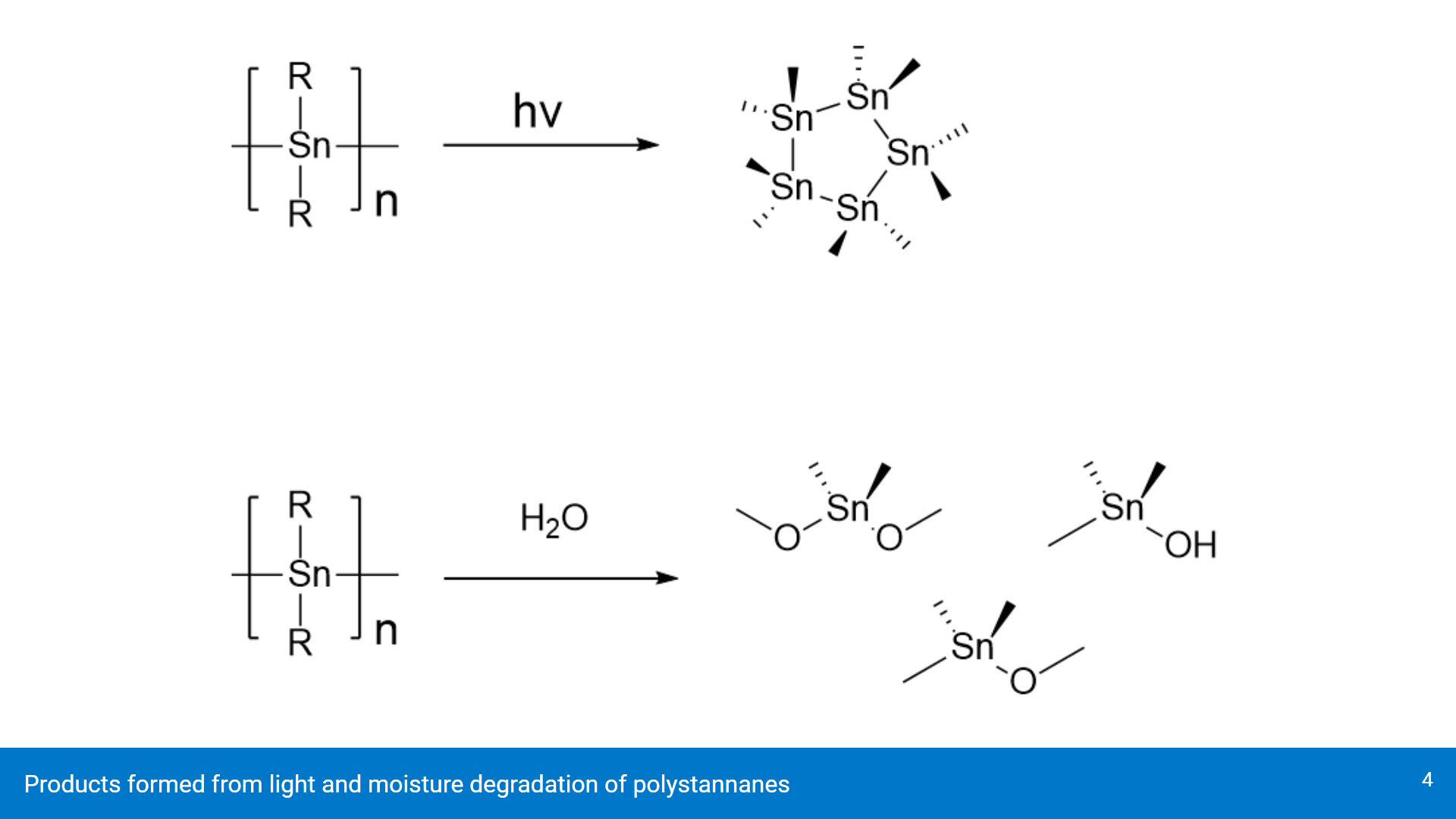
The larger bond dissociation energy is problematic, as the Sn-Sn bonds break with exposure to light (top reaction) and moisture (bottom reaction). Exposure to light causes polystannanes to break down into cyclic stannanes while moisture creates stanoxanes. Both products are not conductive, which is not useful for creating semiconductors for electronics.
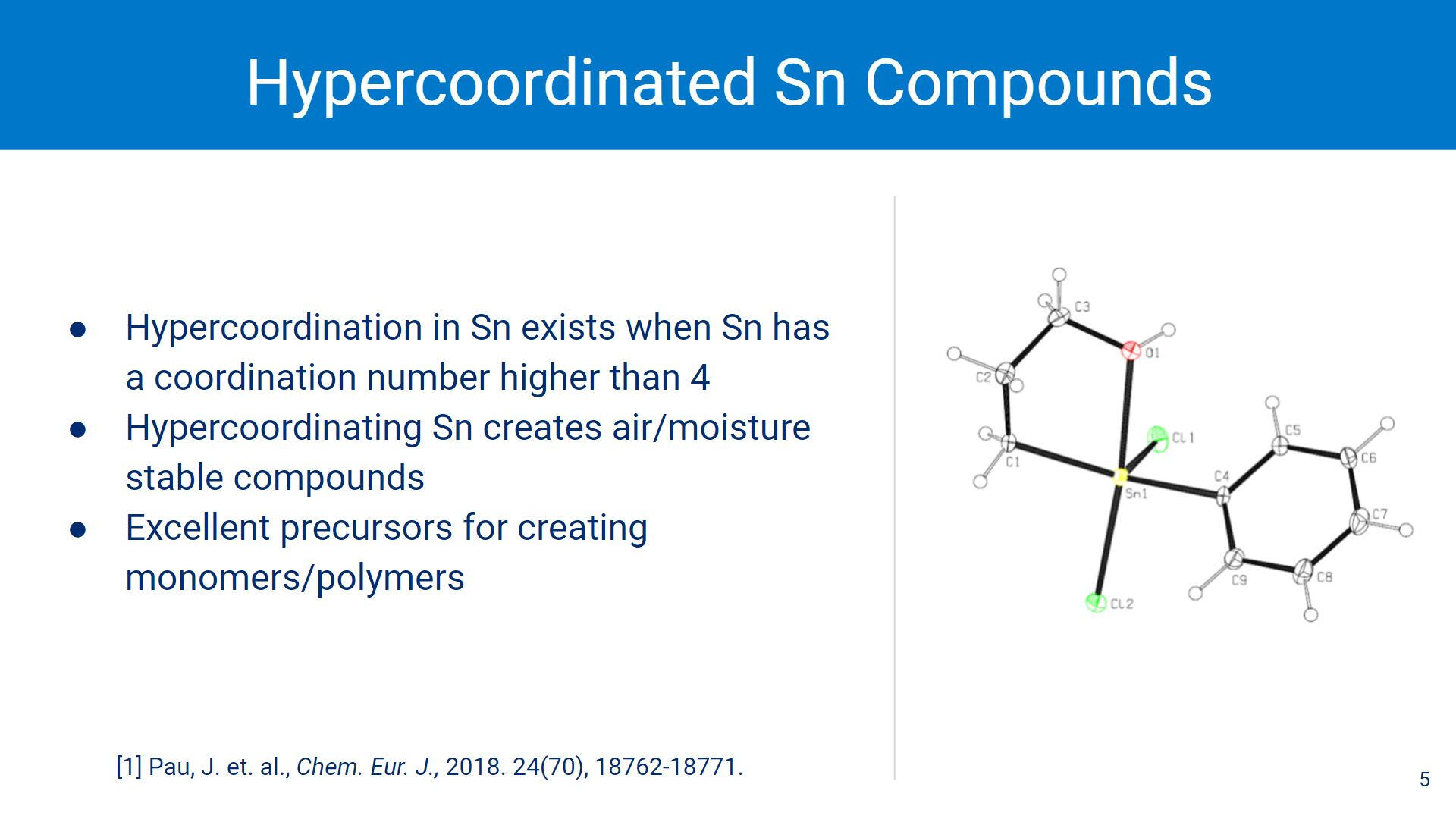
One way to reduce the reactivity of the Sn-Sn bond is by introducing sterics around the Sn centers. 5-coordinate Sn creates air and moisture stable compounds, which is useful for creating polystannane monomers (and eventually polymers).
Reference:
https://doi.org/10.1002/chem.201804145
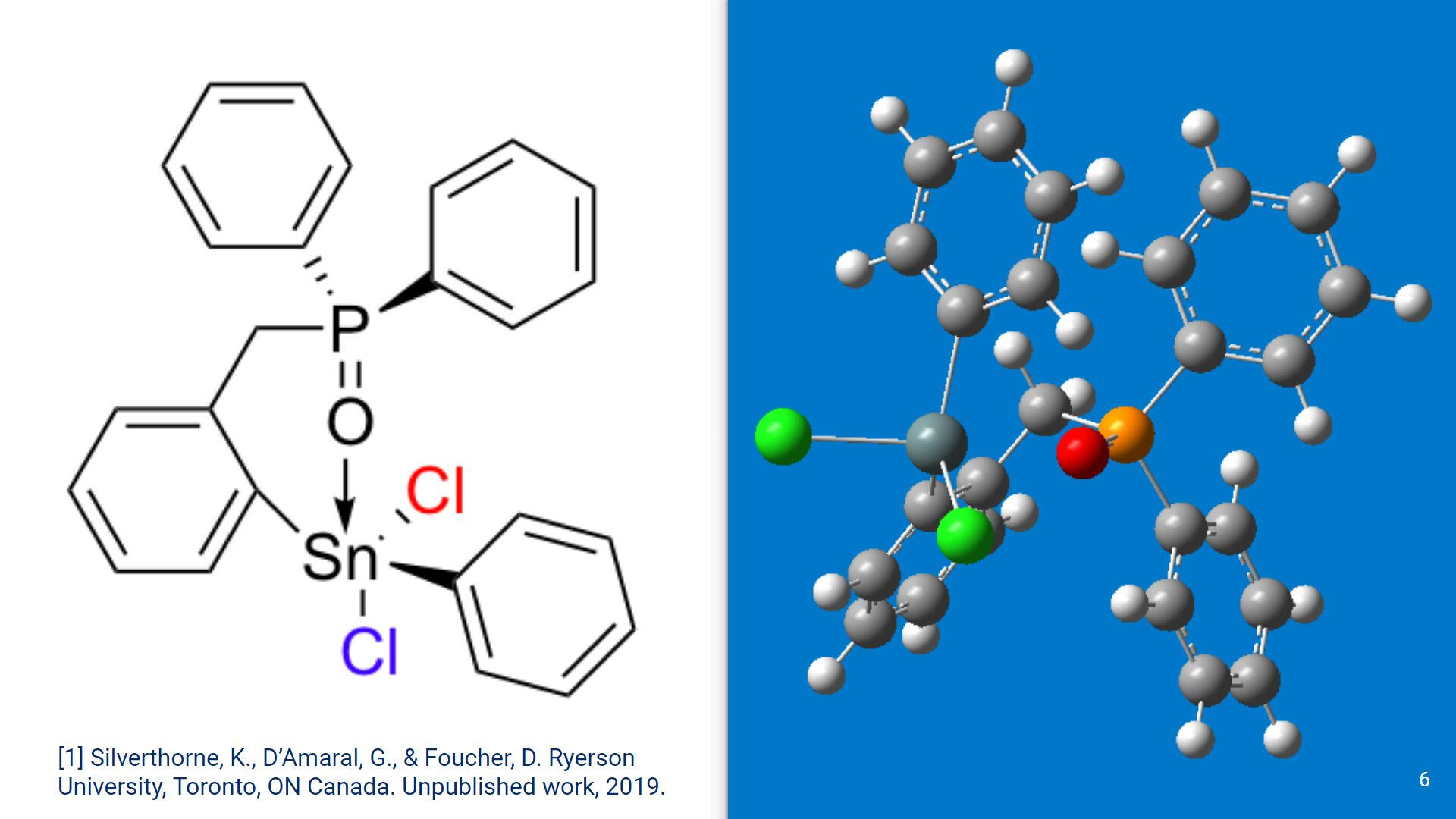
On the left is a graphical representation of a recently synthesized hypercoordinate Sn compound. On the right is a GaussView screenshot of the molecule. This organotin compound differs from previously synthesized organotin species, as it is one of the first systems with a phosphine oxide group coordinating with the Sn center.As of this moment, a solid state structure for this compound has not been found.
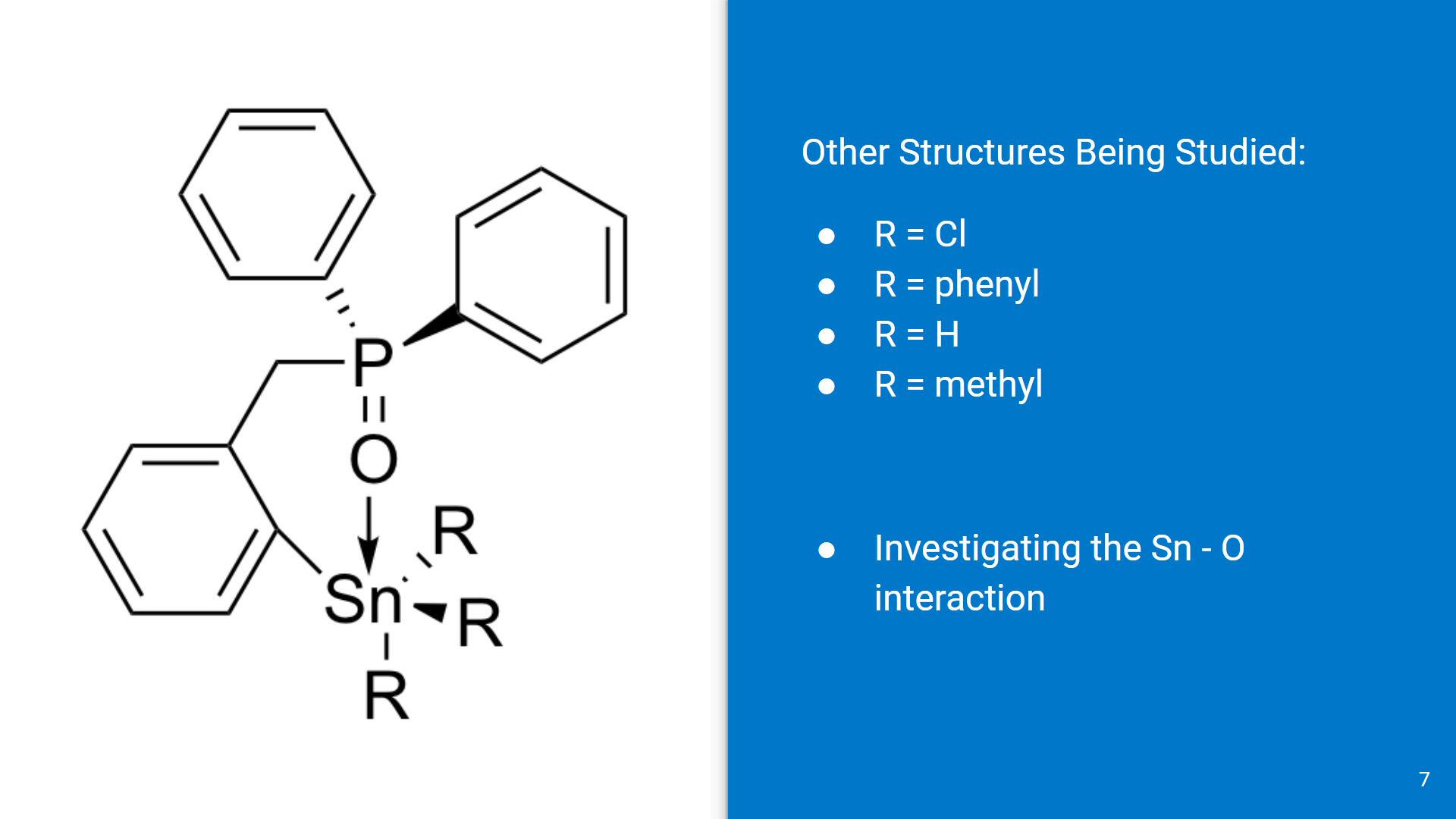
In this ab initio study, the R groups on the Sn center were modified to see how different ligands affect the overall Sn-O interaction.For simplicity, the ligands used were Cl (to model the experimental structure), H (for creating the monomer), phenyl and methyl groups (for geometry and energy comparisons).
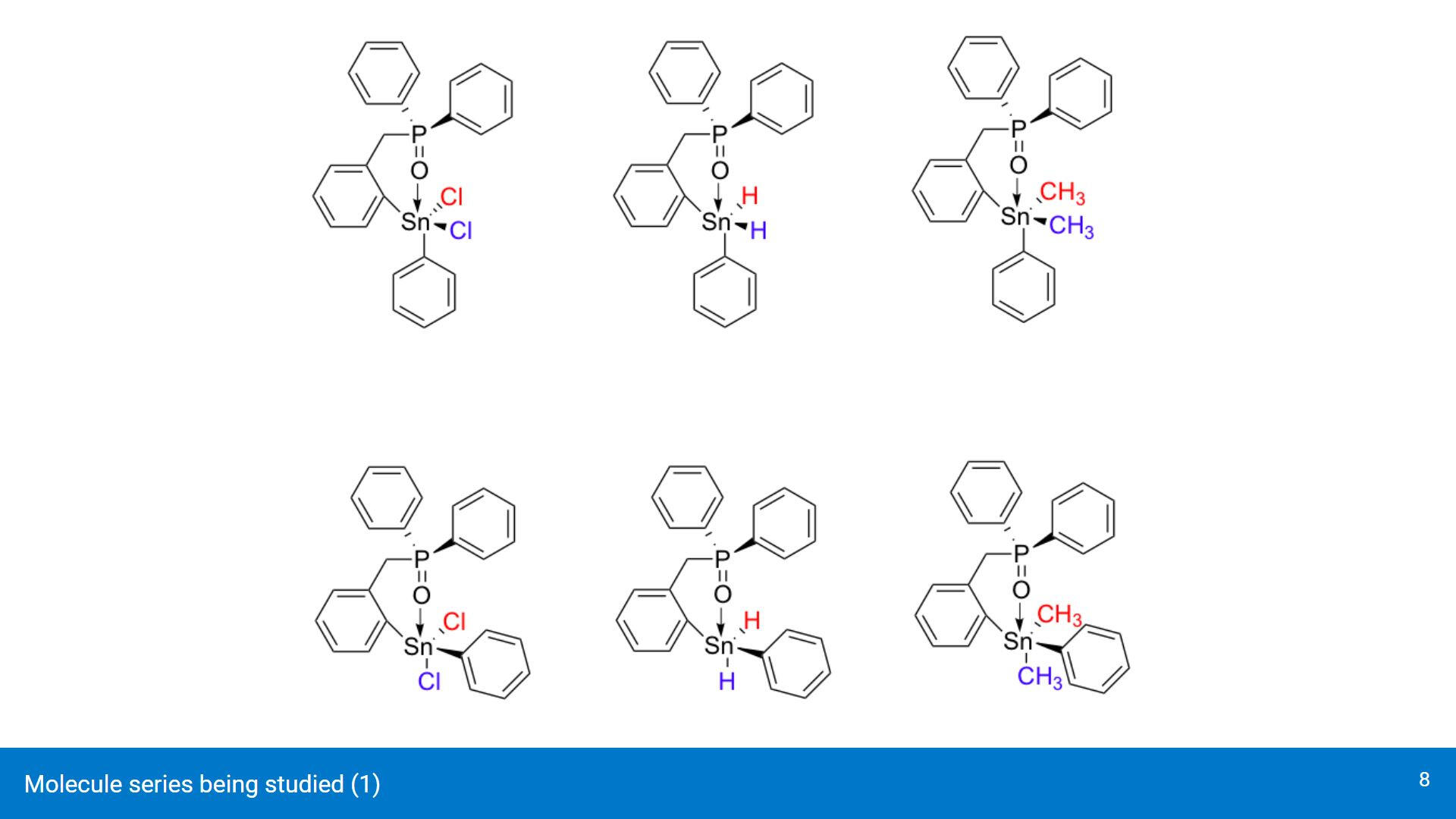
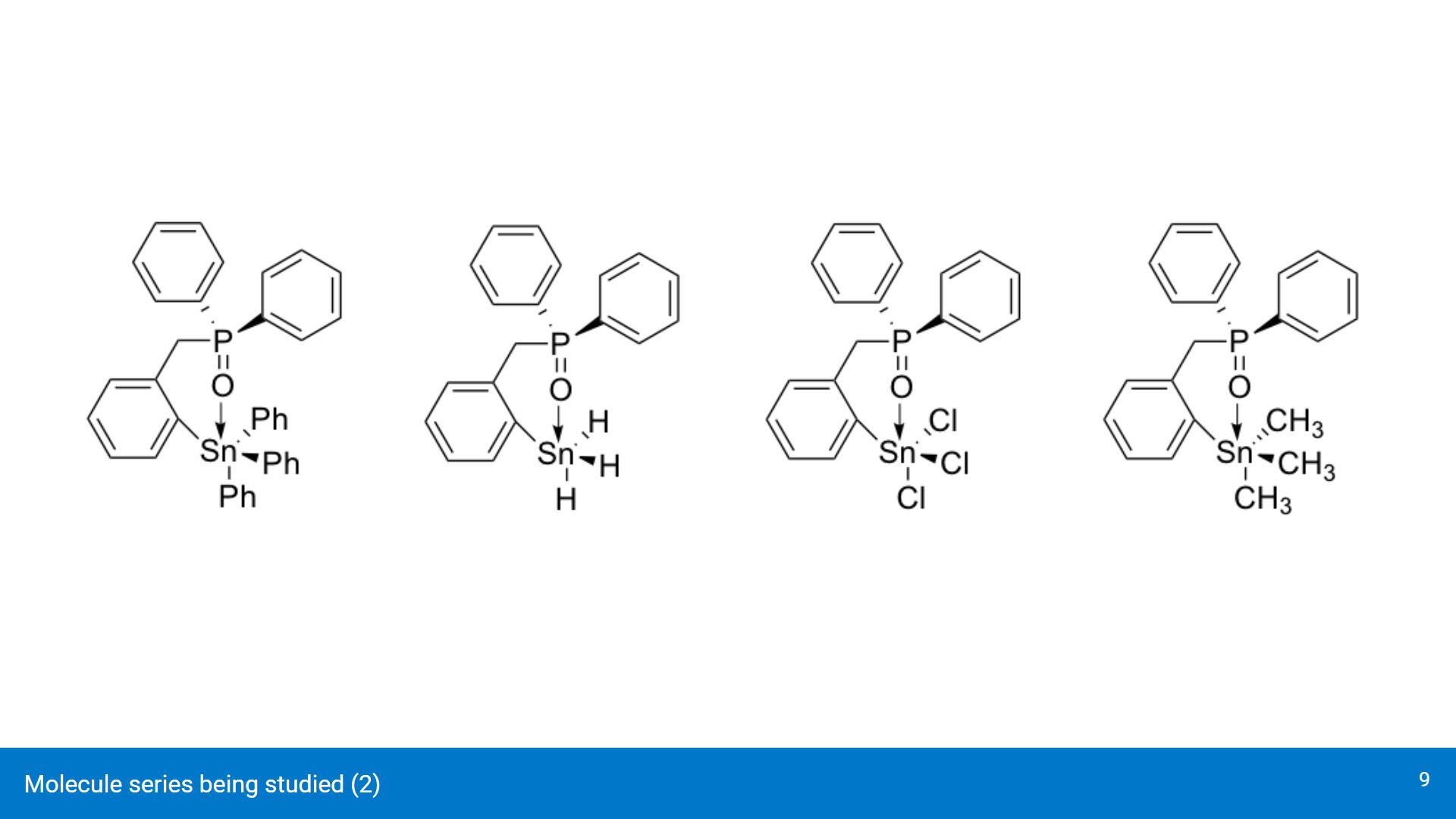
Some of the geometries being analyzed (dichloro, dihydride, dimethyl). Top row contains the cis conformers and bottom row contains the trans conformers.For brevity, the presentation will only cover the dichloro and dihydride geometries.
Though these structures won't be covered in the presentation, the conformers with the same ligands attached to the Sn center will be used mostly to study how they affect the Sn-O interaction.
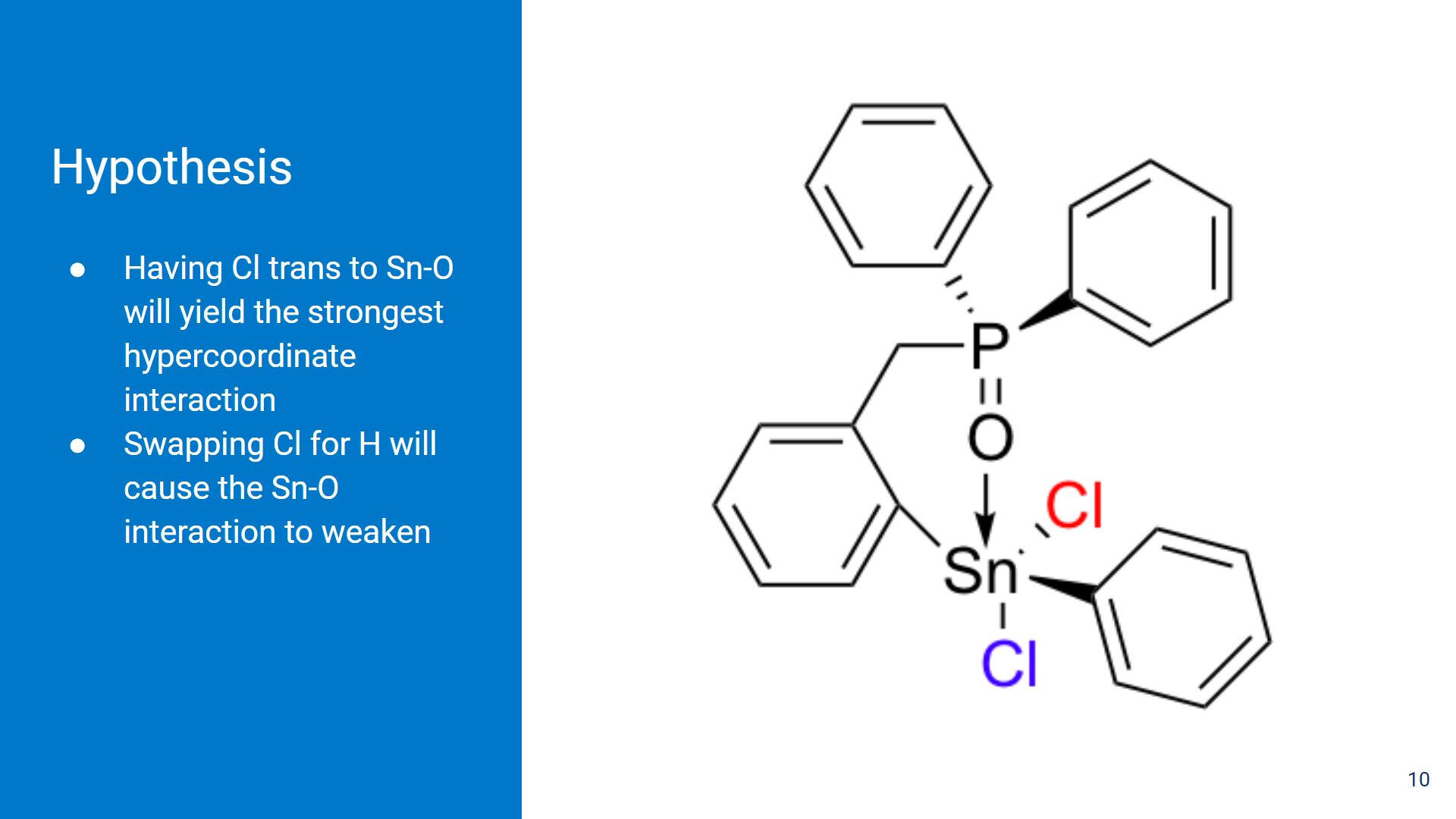
Our hypothesis is that having the Cl ligand trans to the Sn-O interaction will create the strongest interaction, but changing the ligand to something less electron-withdrawing will weaken the interaction.
For our DFT calculations, all of the parameters were kept the exact same for all calculations and done in Gaussian 16. The basis set combination of LANL08d for Sn and 6-31+G(d,p) for C, H, O, Cl and P along with the PBE0 functional + GD3BJ dispersion have been shown in previous DFT studies to be the best for modelling hypercoordinate Sn systems.

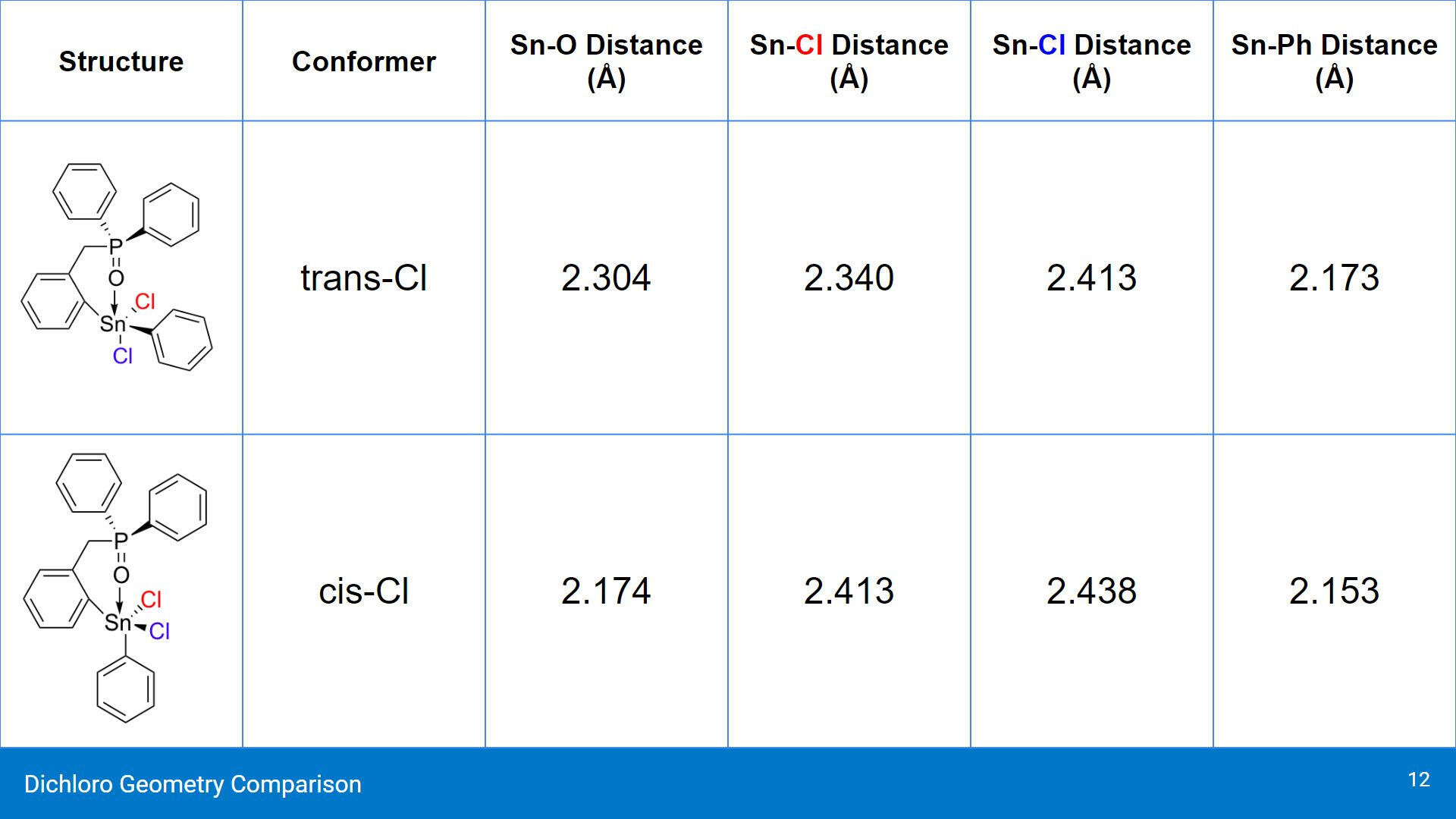
As suspected, the Cl ligand trans to the Sn-O interaction did elongate slightly. What's surprising about these results is that the cis conformer of the dichloro compound showed a smaller Sn-O distance than the trans conformer. In the cis conformer, the Sn-Ph distance also shortened, rather than elongate.
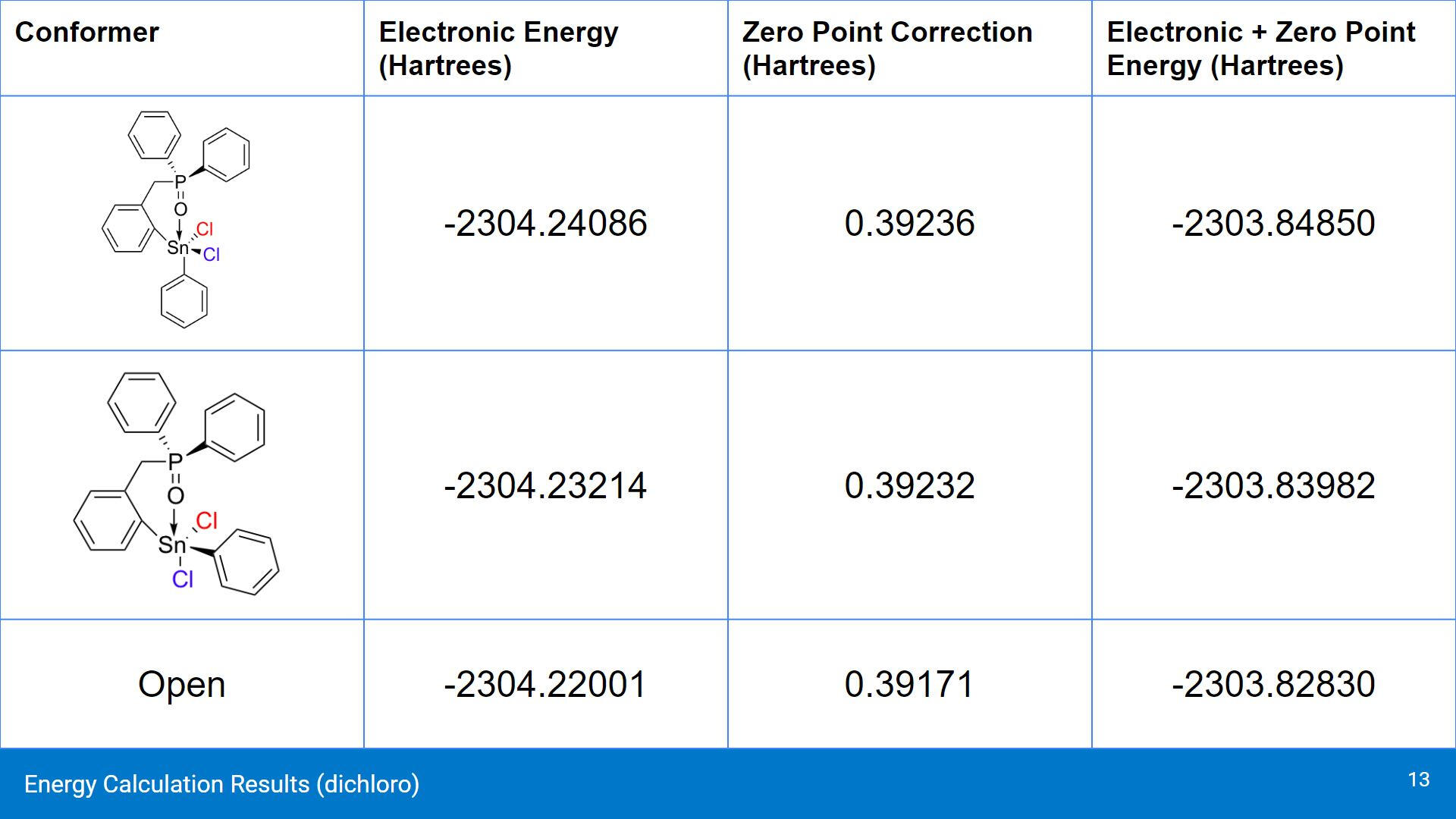
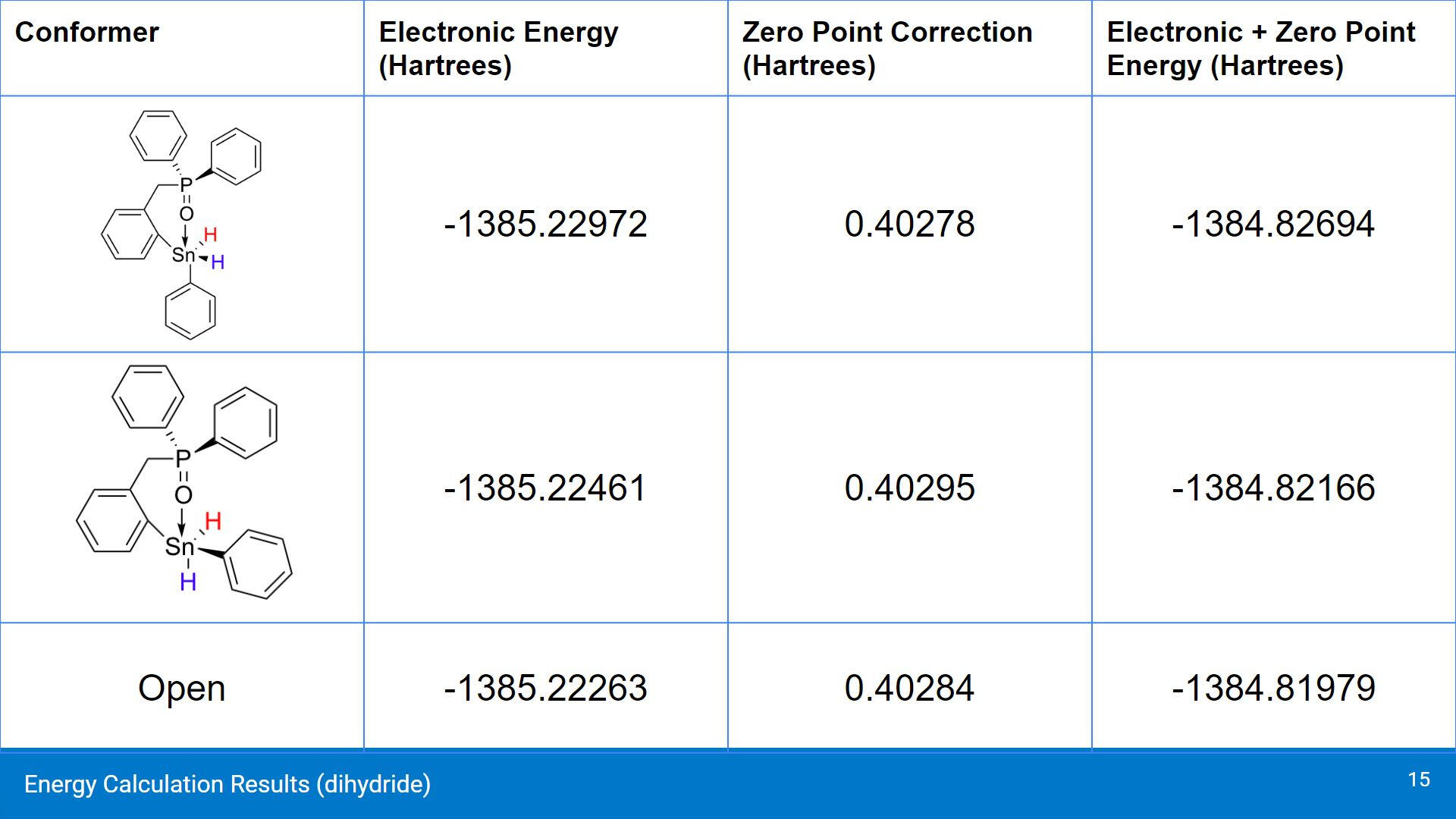
When analyzing the calculated energies, the cis conformer is shown to be the more stable of the 3 conformers. Both the chelating structures of the cis and trans conformers were both predicted to be more stable than the open, non-chelating conformer. In terms of comparing the cis and trans conformers, the energy difference between them is around 0.00868 Hartrees, or 22.785 kJ/mol.
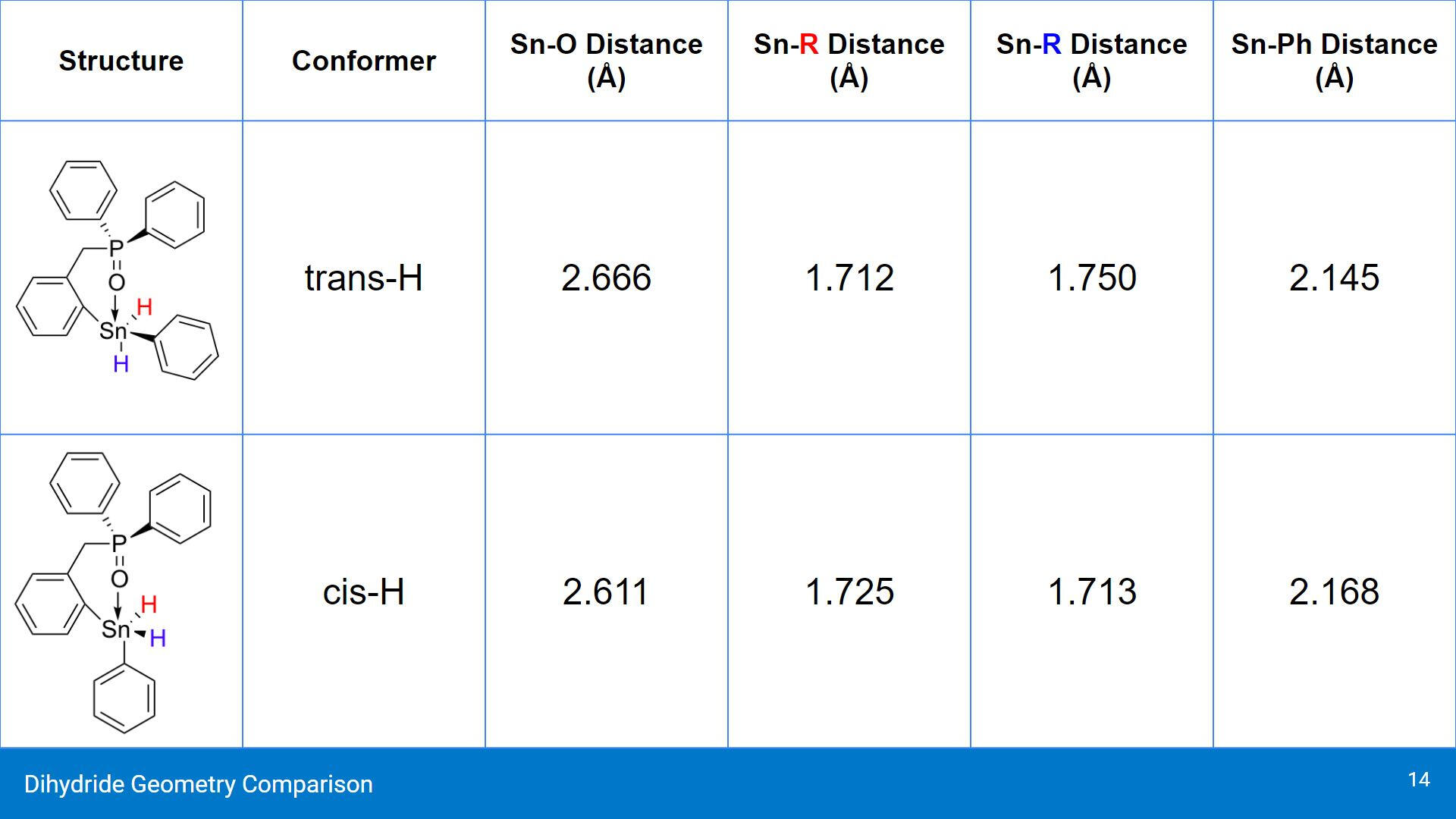
When substituting the Cl ligands for H, there was a significant elongation of the Sn-O interaction in both cases. The cis conformer had a slightly shorter Sn-O distance of 2.611 Å compared to the trans conformer. Something interesting to note is the Sn-Ph distance in the cis conformer is longer than in the trans conformer, which is different than when the compound had Cl ligands.
In terms of their energies, these compounds were predicted to be less stable with the ligand substitution. This is largely due to H being a less electronegative group compared to Cl. H likely does not draw the electron density from the O-P group in a similar way that the Cl ligand has been shown to do. Once again, both the chelating geometries were shown to be more electronically favourable compared to the open, non-chelating conformer. This trend is consistent with the systems that were studied.
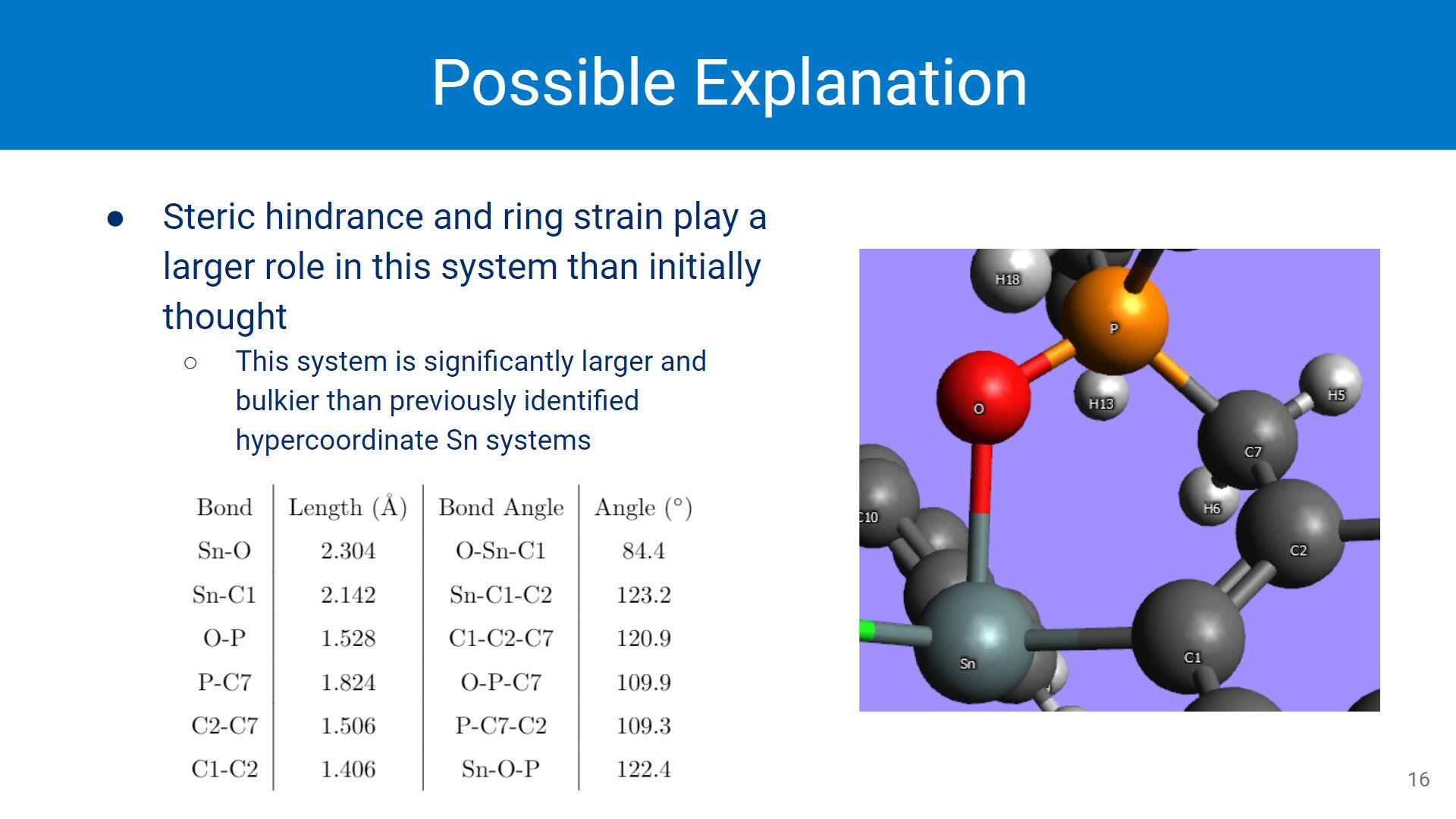
Since this system is one of the first hypercoordinate Sn systems without a solid state structure available, assumptions were made about its behaviours. Previously identified systems would show the Cl ligand trans to the Sn-O or Sn-N interaction, but this is not the case with this system.This system is also significantly larger and bulkier than other previously identified systems. The amount of ring strain created in these systems (depicted) could also have an effect on the overall stability of the conformer.
Despite initial assumptions, the cis conformer of the dichloro organotin showed the strongest Sn-O interaction. Ring strain and overall sterics potentially play a larger role in hypercoordination, though the extent of these roles is still being investigated. Changing the ligands on the Sn center does affect the Sn-O interaction by weakening the interaction.Picture: A very coarse electrostatic potential surface mapped onto the trans-dichloro conformer.


Future work is now focused on investigating the ring strain effect on hypercoordination and looking into the conductivity of these compounds. In terms of the experimental portion, a solid state structure of the compound needs to be identified for comparison to the calculated structures.Picture: One of my longest calculations (triphenyl conformer frequency calculation).
Acknowledgements
This project would not have been as successful if it were not for the following amazing people:Dr. R. Stephen Wylie, my amazing and patient supervisor and professor
Dr. Daniel A. Foucher, my principal investigator and professor
Drs. Stefania Impellizzeri and Marc J. Adler, SOUSCC48 organizers
Kathy, Gloria, Matthew and Rachele from the Foucher and Gossage Groups at Ryerson University
Mokha, Hask, Deidre, Susanna, Tristan, Melissa, Julia, Kelvin, Lavinia, Aviya and Ali, my fellow undergrad chemistry thesis friendsSpecial shoutouts to my friend, Brandon, for making these super fire slides!
Questions?
This serves to replace the question period portion of the presentation. You will be getting a response from me in your email within 48 hours and I will also be posting the answers below for others to view!